A Road Map to Implementation
 The 9th edition of the EU Device Clinical Research Conference will once more dive deep into challenges faced by medical device clinical executives conducting trials for medical products meant to be marketed in Europe. Clinical testing is one of the most critical steps in the route to market, as it involves human subjects, which are protected by a myriad of legal safety measures to ensure the subject’s integrity is safeguarded throughout the study.
The 9th edition of the EU Device Clinical Research Conference will once more dive deep into challenges faced by medical device clinical executives conducting trials for medical products meant to be marketed in Europe. Clinical testing is one of the most critical steps in the route to market, as it involves human subjects, which are protected by a myriad of legal safety measures to ensure the subject’s integrity is safeguarded throughout the study.
Looking specifically at the main concerns for the audience this year, there is a lot to say about regulations relevant to clinical practice, as the European Union progressively moves into a new set of rules, the EU Medical Device Regulation. So far, European member states would implement directives issued by the EU Commission into local legislation, whereas now with the MDR all member states will align with a single set of rules, implemented seamlessly in all member states. While the harmonization of requirements is greatly appreciated, the complexity of the new rules leave the industry mesmerized. The MDR was adopted late May 2016, but the final text has not been published yet, creating much anxiety in the industry, trying to figure out what they should do to update clinical strategies. The focus is mainly set on more safety for patients, more safety for practitioners, more clinical data to prove product safety and effectiveness, more scrutiny from authorities, more clinical testing altogether, more more more!
The following article, written by Gert Bos, Executive Director & Partner at Qserve and distinguished presenter at the 9th Annual EU Device Clinical Research Conference outlines a road map to implementation.
Following the approvals on the legislative proposals on both EU-MDR and EU-IVDR, the summer period has been utilized to translate the final draft that was agreed upon in principle into all EU languages. In August, a revised consolidated draft was released, and another version with first minor tweaks was circulated in the middle of October. The next steps was to get feedback from the EU member states on the translations and legal correctness of the final draft texts. The period of commenting has just ended now, and work is in progress to capture all and work resolve any discrepancies. So, progress is being made, and publication is getting closer to reality. Last estimates are finding Q2 2017 as likely period for formal publication, and entry into force – kicking off the transition period of 3 years for EU-MDR and 5 years for EU-IVDR.
The new Regulations will shift the landscape and will bring the safety and performance of medical devices to the next level, that also has been strongly requested by authorities, patient groups and the general public. And not only that, it also has a system of additional implementing and delegated acts that will allow the legislators to over time adjust the requirements to the state of technology and the ongoing healthcare improvements. As such it truly is built to last with future proof mechanisms in place. Another such mechanism is the system of so-called ‘common specifications’, detailed descriptions of requirements and policies that can be added over time to instruct all stakeholders on minimum expectations in terms of clinical safety, clinical performance, post market surveillance and follow up of patients in specific cases in post market clinical follow up protocols.
Meanwhile we see the early implementation already taking off. Medical Device Expert Group meetings make place for new ad-hoc meetings between the competent authorities and notified bodies, where the common interpretations and expectations from the member states are shared with the notified bodies. Notified bodies have meanwhile strengthened their coordination of finding joint interpretations on ongoing matters of early interpretation. Also continuing is the re-designation process that was initiated in 2013, to lift notified bodies in the direction of the new requirements. Strong joint assessments clearly improve the quality and consistency of work from notified bodies, but also continue to see notified bodies being reduced in scope, or even being closed altogether. Current number of remaining notified bodies is 58, whilst not all have gone through the new strict evaluation process.
As a result, from the stricter control on notified bodies, and the push away from conformity assessment to compliance inspection, typically the number of deficiencies identified in dossier reviews and audits go up, and also the level attributed to them is increasing, so numbers of major non-conformities found in audits see a dramatic increase. Serious follow up and improvements are required, which might be challenging, but on the other hand are also helpful to get into early compliance with the upcoming legislation.
Companies slowly move into transitioning phase, raising internal awareness and commitment, and carefully increasing budget for regulatory compliance. Gap assessments and portfolio analysis should be starting by now. If you wish to verify what stage you are in compared to others working on their EU-MDR implementation, you should check out our EU MDR First Aid app. (available Januari 2017)
A lot of work at hand for all involved, as we realize the impact might be even bigger than anticipated. Especially now that we see notified bodies tightening the reigns on the clinical evaluation requirements following the release of MEDDEV 2.7.1 rev.4, companies increasingly realize their clinical evidence might resemble too much the card house of stapled equivalence contributions. Time is there to urgently collect clinical data on one’s own products in PMCF mode.
With the limited time available for transitioning, realizing the last year might be needed for notified body assessments, and corrective actions on non-conformities found, the time left to get clinical data in, or to make for example changes in the supply chain, select a new notified body, or other time consuming changes, is becoming critical.
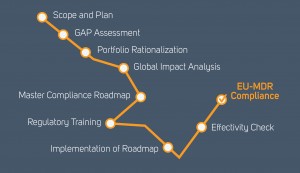
If you wish to learn more on the details of the implementation, in an informal workshop setting, where you can also discuss with your peers, please check out the modular training that will start in Amsterdam early in the new year. Getting in to selected workshops, or getting a company seat so that different colleagues will be able to visit different topics, will greatly enhance your readiness and knowledge moving into the transition. More details on “Where are you on the road to EU MDR Compliance”
Dr. Gert W. Bos, Executive Director & Partner at Qserve.
 Gert has 21 years of experience in life sciences (devices and pharma), in university, industry as well as in four Notified Bodies. His last notified body roles were Head of Regulatory and Clinical Affairs of BSI Medtech and Head of Notified Body at BSI-Germany (NB0535). He has been president of the Notified Body association TEAM-NB, and vice-chair of the Medical Notified Body forum NB-Med in Brussels. In these roles for many years he represented notified bodies in a.o. the Clinical Investigation and Evaluation Group (CIE), Medical Device Expert Group (MDEG) and the MDEG workgroups on animal tissue, on MRA’s, e-labeling, EUDAMED and on IVD’s, as well as the IMDRF workgroup on Regulated Product Submissions and Table of Content. In addition, he served as NB-representative at EMA/CAT and Medical Device Collaboration group. He is a founding board member of the Dutch RAPS chapter.
Gert has 21 years of experience in life sciences (devices and pharma), in university, industry as well as in four Notified Bodies. His last notified body roles were Head of Regulatory and Clinical Affairs of BSI Medtech and Head of Notified Body at BSI-Germany (NB0535). He has been president of the Notified Body association TEAM-NB, and vice-chair of the Medical Notified Body forum NB-Med in Brussels. In these roles for many years he represented notified bodies in a.o. the Clinical Investigation and Evaluation Group (CIE), Medical Device Expert Group (MDEG) and the MDEG workgroups on animal tissue, on MRA’s, e-labeling, EUDAMED and on IVD’s, as well as the IMDRF workgroup on Regulated Product Submissions and Table of Content. In addition, he served as NB-representative at EMA/CAT and Medical Device Collaboration group. He is a founding board member of the Dutch RAPS chapter.




