Aligning with MEDDEV 2.7.1 and the MDR
The newest revision has defined, clarified and tightened guidance on clinical evidence requirements for unapproved technologies, as well as products that have been on the market for an extended period. It’s much clearer in the route that manufacturers have to take to bring their clinical evaluations to fulfillment. It’s also much clearer on what notified bodies should be looking for.
“It was written bearing in mind the changes through the MDR, and has very much tightened up on clinical.” Explained Anwen Evans, Certificate Decision Maker, Underwriters Laboratories in an interview with Q1 Productions. “The MEDDEV reflects the direction that the MDR is going in. Which is going to be a big help to manufacturers.” A lot of changes have been set to ensure more safety for patients and practitioners, more clinical data to prove product safety and effectiveness, more scrutiny from authorities, and more clinical testing altogether.
Integration
The recently published MEDDEV 2.7.1 rev 4 is currently active with no transition period, and moving forward, notified bodies will seek to review all device’s CERs, regardless of when the device was approved and placed on the market. Manufacturers must be transparent and detailed about the methods used and steps taken during the clinical evaluation process and provide detailed data analysis, process logs and lists of references. With no current determination on the level of acceptable data, manufacturers should already be analyzing product portfolios, conducting impact assessments and collecting as much clinical data as possible to satisfy future requirements.
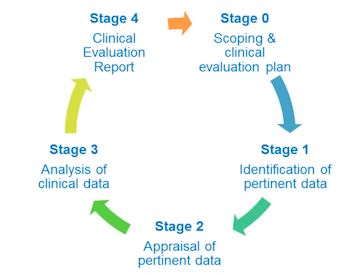 There are five stages from the scoping and clinical evaluation plan to finalization of the CER.
There are five stages from the scoping and clinical evaluation plan to finalization of the CER.
Stage 0- Scoping & Clinical Evaluation Plan
Stage 1- Identification of Pertinent Data
Stage 2- Appraisal of Pertinent Data
Stage 3- Analysis of Clinical Data
Stage 4- Clinical Evaluation Report
We talked to Anne Le Rouzo West Coast Regional Director & Certification Project Manager / Lead Auditor LNE / G-MED North America, Inc. about developing evaluation reports throughout the product life cycle, details on inclusion/exclusion criteria & usability requirements, new and revised definitions. Watch her hour-long webinar here.
Impact
The overall concept of clinical research does not change; the manufacturer will still use clinical data to demonstrate compliance. Data is still analyzed based off premarket and postmarket investigations. The impact comes into effect as the different level of details, systems and timing.
The product most impacted by the changes are high-risk devices. The CER for high-risk are more detailed and needs more data than before. High-risk and innovative products now have to update their CER annually and anytime new data is collected. Even high-risk devices that were considered equivalent must be updated. Equivalent devices are newly defined as almost identical products. There is great detail in MEDDEV rev 4, Appendix 1, defining what equivalence now means. Clinical, technical and biological characteristics must be considered for the demonstration of equivalence.
The second most significant impact is that it is no longer going to be a one-time update, but a continual update. Starting in the design stage, clinical evaluations are required. It is an ongoing update throughout the entire life-cycle of a product, and all documentation must match. While high-risk devices must be submitted every year, manufacturers must justify the frequency of updates for lower risk devices. These justifications must consider design changes, risk, and new data. They should do this every two to five years.
The third impact is that the summary of clinical data will be publically available. There will be a summary of clinical data available contrasted to an assessment of complaints and the volume of sales. A postmarked surveillance report updated annually with key information of what the manufacturers knows about the safety of the product and the performance of the product on an ongoing bases. It is also required to include methods and steps taken to collect data. The CER will have a log of how and when the evaluation was performed. Therefore, it will be comparable with what competitors have in the market, so it will impact both the visibility of the product and their problems compared with the others. Not only effecting transparency of sales, but also providing journalists with and abundance of information for potential public-affairs issues such as recalls.
Implementation
Although there is no official transition period to the guidance, manufacturers should consider if they can incorporate their implementation plans directly into the MDR compliance. In some cases they can, but some products have only until the end of the year to submit their CERs. Any documents that are to be reviewed by a notified body will have to be updated separately to revision 4. Those that can be updated a little later can wait until more information is available, and update to the draft of the future MDR.
With the transition into the new Medical Device Regulation, device manufacturers are required to obtain the new CE mark for existing as well as new products. As there are no grandfathering clauses enabling a shift to the new CE mark outside of clinical testing, legacy products that have been on the market for years also need to undergo further testing under the new rules. Clinical teams are therefore currently asked to assess legacy device trials feasibility, with a focus on the overall time and cost to forecast, in order for the corporation to make enlightened decisions about legacy product lines to keep on the market or terminate. “At this moment the lack of clinical data from manufacturers that are early on in the interpretation indicates that they might have to say farewell to using up to 50 percent of their portfolio” explained Gert Bos, Advisor CIE Taskforce on MEDDEV 2.7.1 to Q1 Productions in an interview.
The CE mark within this legislation is a game changer. It’s not just a brief update of existing documentation to be able to show compliance. It’s a new concept altogether, with new thinking, and enhanced expectations. It is stricter, and for products to stay on the market in the new regime, especially for higher risk products, there needs to be sufficient clinical data. There is an emphasis on the word sufficient. It has not been defined yet, so we are all trying to figure out what that means. One thing is certain, it is much more than ever before.
“Manufacturers should be almost ready for the changes, but in reality, the majority have barely started clinical evaluation reports compliance with the new guidance,” said Gert Bos. Staying up to date on clinical news is more important than ever. That’s why, for the 9th year in a row, clinical executives will meet for the EU Device Clinical Research Conference in Berlin on January 23-24. This conference will continue to provide executives with an unparalleled educational opportunity, through a distinct mix of session formats. Traditional presentations delivered by high-level speakers from the industry, competent authorities and notified bodies will be followed by interactive group discussions as well as engaging workshops focusing on the practical aspects of new rules implementation and more. Further topics of the utmost importance will also be debated, such as the pros and cons of outsourcing trial operations, meeting ethics committees’ expectations and clarifying clinical evidence demands for reimbursement. With such a variety of formats, speakers, and topics, the 9th Annual Q1 European Medical Device Clinical Research conference is a must-attend for forward-thinking clinical affairs professionals.




